
Molecular packing determines singlet exciton fission in organic semiconductors
K. Kolata, T. Breuer, G. Witte, S. Chatterjee
ACS Nano 8 (2014) 7377
The optimum performance of conventional photovoltaics is constrained by the Shockley-Queisser limit; about 30 % efficiency for silicon devices. Carrier multiplication by singlet-exciton fission in molecular semiconductors may enable enhanced efficiencies. Experiments shed light on carrier multiplication dynamics and reveal their relation to the molecular arrangement and consequently the intermolecular interaction in a model single crystalline system.

Energy level scheme and underlying relaxation mechanisms implemented in a rate-equation model. Solid blue arrows represent radiative recombination; the dashed arrows non-radiative relaxation. Red arrows indicate induced absorption. The inset shows the photoluminescence spectrum of the sample.
Singlet-exciton fission describes the conversion of a singlet exciton into two triplet excitons. In conventional photovoltaics, absorbed photons are converted into excitons that dissociate into a net current and excess energy is dissipated as heat. Carrier multiplication utilizes this excess energy to generate additional excited carriers. When the photon energy for generation of singlet excitons is about double or more than for triplet excitons, the excitation may be distributed to two triplets excitons. These are parity-forbidden for individual molecules and cannot be excited directly by individual photons. However, once triplet states are populated, they may contribute to charge generation and hence increase the quantum efficiency, eventually even above unity.
Experimental investigation by time- and polarization-resolved pump-probe measurements reveals a correlation between singlet-exciton fission and the molecular arrangement. Exploiting the specific epitaxial growth relations of the prototypical organic semiconductor perfluoropentacene (C22F14, PFP) on KCl(100) and NaF(100) substrates renders it possible to independently address all three crystal axes by linearly polarized light. The observed spectral signatures are highly anisotropic due to a strong uniaxial delocalization of the excitations which, in turn, leads to weakened molecular selection rules. The pronounced slip stacking of the molecules along the b-axis enhances the intermolecular coupling, accompanied by delocalization of singlet excitons and direct coupling to the correlated triplet pair 1(TT). These findings significantly enhance the fundamental understanding of the relation between molecular arrangement and electronic response laying the foundation for future high-performance organic electro-optical devices.
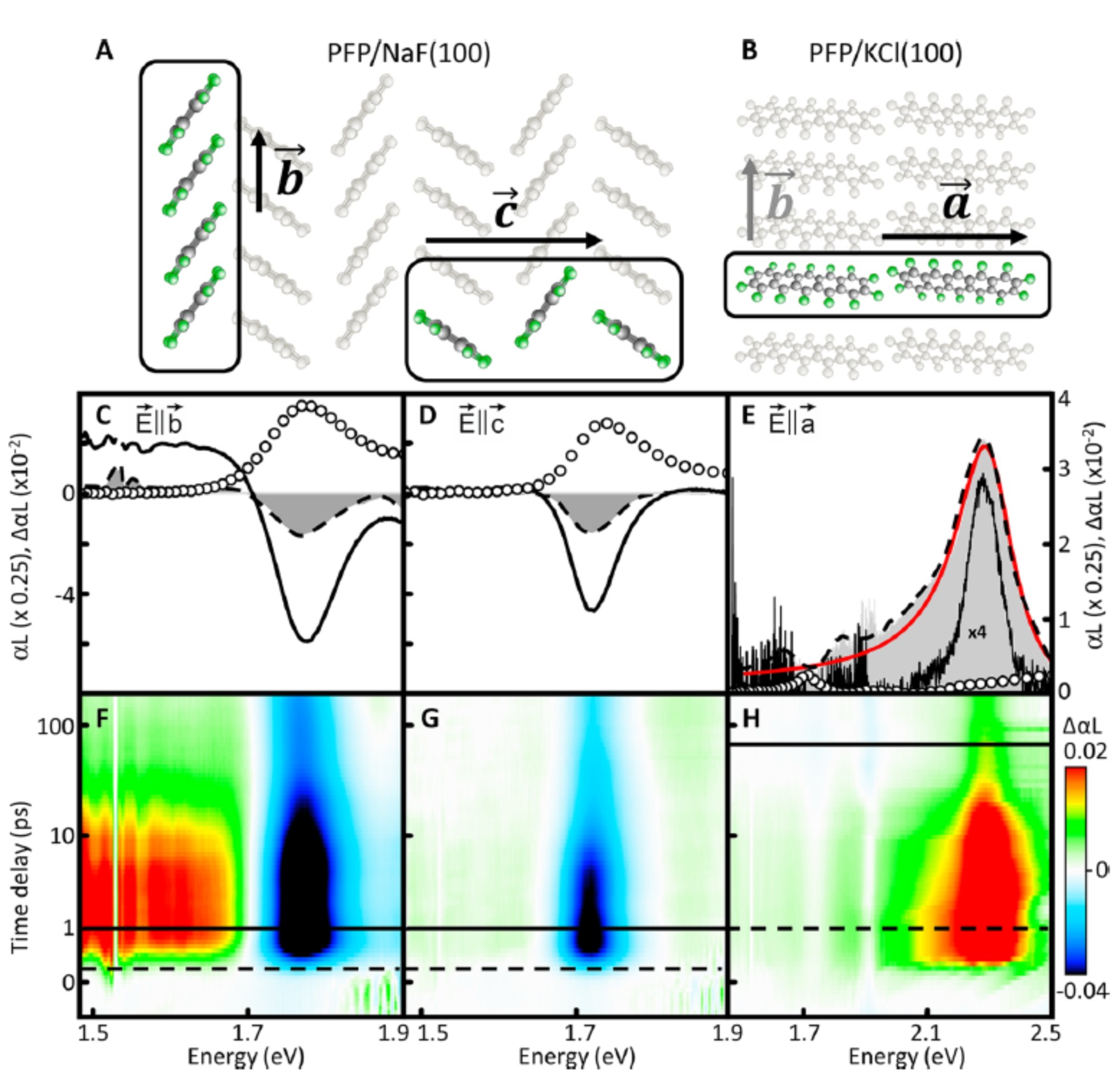
Correlation of packing scheme (A–B) and optical response (C–H) along all crystal axes. Linear absorption (circles) and corresponding differential absorption spectra at the time delays of 300 fs (dashed) and 1 ps (solid), as well as induced absorption and a Fano fit (red) (c⃗-axis) at 1 ps (dashed) and 90 ps (solid). The time evoluation of the differential absorption are shown on a nonlinear time scale.